宏基因组测序技术已经广泛应用于微生物的研究当中,如何利用测序短序列重构出质量可靠的微生物基因组,对于理解微生物群落和下游功能分析至关重要。尽管近些年来组装算法取得了很大进展,但是由于宏基因组数据的固有复杂性,错误在宏基因组组装结果中仍然普遍存在,包括由于同一基因组内的重复序列或不同基因组之间共享的保守序列引起的基因组内或者基因组间拼接错误。现有评估宏基因组组装质量的方法大都依赖参考基因组,仅适用于真实环境中已知的一小部分微生物,这显然不适用于微生物组数据中大量的未知物种。因此,亟需针对基因组组装结果开发不依赖参考基因组的质量评估以及错误矫正方法。
复旦大学类脑智能科学与技术研究院赵兴明教授团队提出了一种无参的宏基因组组装错误识别以及校正工具metaMIC,能够精确定位组装重叠群(contig)上可能的错误区域。2022年11月4日,该研究成果以metaMIC:Reference-free Misassembly Identification and Correction of de novo metagenomic assemblies为题,发表在Genome Biology期刊。
metaMIC不仅可以适用于宏基因组组装错误的识别,同样也适用于单基因组组装质量的评估。metaMIC是一种基于机器学习的评估组装重叠群质量的方法,其主要步骤包括特征提取、识别存在组装错误的序列以及定位组装错误位点并校正三个步骤(图1)。
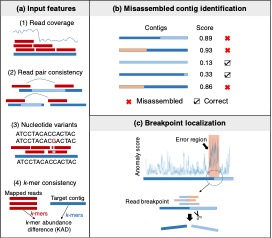
图1: metaMIC框架
metaMIC在多个模拟宏基因组数据以及单基因组测序数据上均取得了良好的性能,其相比于已有工具能够以更高的精度识别存在组装错误的序列,并且大概~71-86%的组装错误断点能够以500bp的误差被精确定位。
最后,研究团队将metaMIC应用在两个人类肠道宏基因组测序队列中识别到了大约3%的组装序列存在拼接错误。通过比较metaMIC校正前后的分箱(binning)结果,发现经过metaMIC组装矫正后能够恢复更多的高质量基因组。由于PacBio三代测序的长读段能够跨越重复序列区域(造成组装错误的主要原因),研究团队进一步以三代测序的组装结果作为金标准来验证预测的二代测序组装结果中的错误,进一步证实了metaMIC在真实宏基因组数据上的适用性。
复旦大学类脑智能科学与技术研究院生物医学AI团队的博士生赖森莹是本研究的第一作者,赵兴明教授、Luis Pedro Coelho青年研究员以及华中科技大学的陈卫华教授为本论文的共同通讯作者。近年来,该团队在微生物领域进行了不断探索,围绕宏基因组组装、物种识别到下游分析已开展了一系列工作,相关工作包括宏基因组分箱算法SemiBin (Nature Communication, 2022)、全球微生物基因目录GMGC (Nature, 2021),建立了GMrepo (Nucleic Acids Research, 2019, 2020), mMGE (Nucleic Acids Research, 2021)和mBodyMap (Nucleic Acids Research, 2022)等数据库。
原文链接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02810-y




