近日,复旦大学附属儿科医院感染传染肝病科研究团队发现并报道了一种新的遗传性肝病。这一成果由复旦大学附属儿科医院、复旦大学附属金山医院、中国科学院深圳先进技术研究院、复旦大学附属华山医院、上海交通大学附属仁济医院、复旦大学生物医学研究院及奥地利格拉茨大学医学院病理系多家单位合作完成,并于7月31日在线发表于BMJ旗下的全球知名遗传病杂志Journal of Medical Genetics。该研究揭示了一种新的遗传性肝病,致病基因为ZFYVE19,临床表现为先天性肝纤维化、硬化性胆管炎和高GGT胆汁淤积症。

图1. 论文《ZFYVE19缺陷相关先天性肝纤维化、硬化性胆管炎和高GGT胆汁淤积症》由Journal of Medical Genetics在线发表

图2. 部分上海研究团队成员:左侧,从前往后依次为栾维莎博士后(共同第一作者)、郝陈指博士(共同第一作者)、陆怡医生、谢新宝博士、库尔班江博士、李忠蝶硕士生、丘倚灵博士后、卢仕琪硕士、王建设医生(通信作者);右侧上,肝病科病理顾问Alexander Knisely教授;右侧下,黎佳琪硕士(共同第一作者)。
随着经济社会的日益发展,儿童肝病的疾病谱在数十年间发生了巨大转变,由原来以传染性肝炎为主,逐渐转变为以遗传性肝病等罕见病为主。作为国内最大的儿童肝病诊治和研究中心,复旦大学附属儿科医院肝病科在多年的临床工作中积累了丰富经验,并在立足于临床病人的科研中收获了诸多成果。2017年,王建设教授领衔的肝病团队鉴定了MYO5B缺陷引起的低GGT胆汁淤积症谱系,被国际上公认为家族性胆汁淤积症6型(图3)。今年3月,该团队又在国际上率先报道了一组由USP53缺陷引起低GGT胆汁淤积症谱系,详细阐述了该病的临床特征及病理特征,并揭示了这一新基因与已知低GGT胆汁淤积症致病基因TJP2的潜在关联(图4)。而近期报道的ZFYVE19则是全球首次关于该基因致病的报道,临床上表现为先天性肝纤维化、硬化性胆管炎和高GGT胆汁淤积症。

图3. MYO5B缺陷导致低GGT胆汁淤积症疾病谱系,2017年发表于Hepatology

图4. USP53缺陷导致低GGT肝内胆汁淤积症的临床特点、病理特征及超微结构,2020年发表于Liver International
探索真理的道路从来不是一帆风顺。从初次通过二代测序方法从病人中筛选出ZFYVE19,到研究成果被认可并发表,历时六年,在多方协作、共同努力下,经历了多位研究团队成员的接力攻关。
为了寻找致病证据,团队研究人员积极收集更多病例、随访病人,提供具有说服力的家系证据;与华山医院、仁济医院合作,收集病人肝移植术中的组织样本进行研究,证明在病人中该蛋白表达缺失(图5);与奥地利格拉茨大学医学院病理系合作,分析研究病人肝组织的病理特征,从病理特征推测此病应该是纤毛病(图6),终于找到进一步深入研究的线索;与中科院深圳先进技术研究院合作,通过细胞系模型和病人来源的诱导干细胞实验证实ZFYVE19变异影响纤毛形成,首次明确该基因与纤毛形成的直接相关性(图7);与复旦大学生物医学研究院合作,发现ZFYVE19基因的数据库注释存在缺陷,从而导致这一基因在测序分析中容易被忽略。研究人员通过该基因转录和蛋白翻译方面进行深入研究,对该基因的翻译起始位点进行了新的注释,克服了注释错误对研究造成了多重壁垒(图8)。
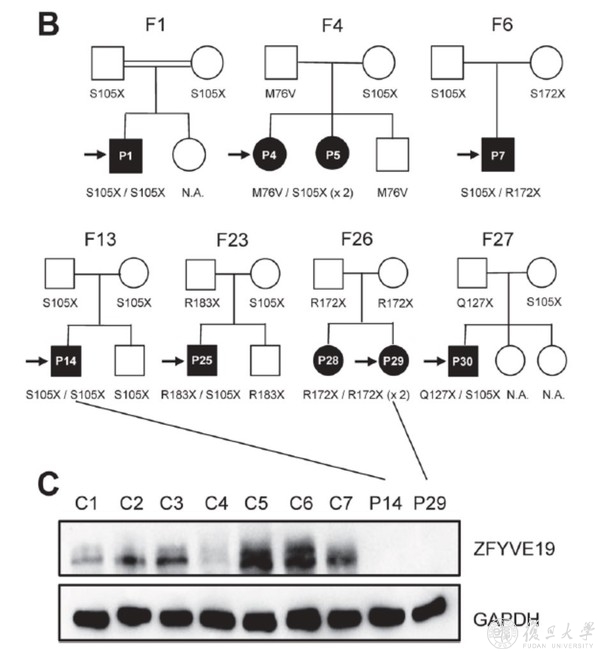
图5. ZFYVE19缺陷病人家系图显示符合常染色体隐性遗传,病人肝组织中ZFYVE19蛋白缺失。
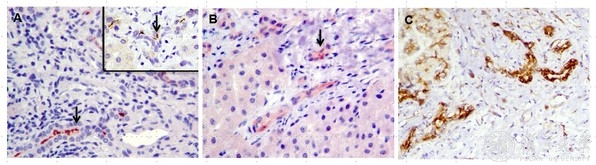
图6. 病人肝脏组织病理中纤毛蛋白表达异常,提示纤毛病可能
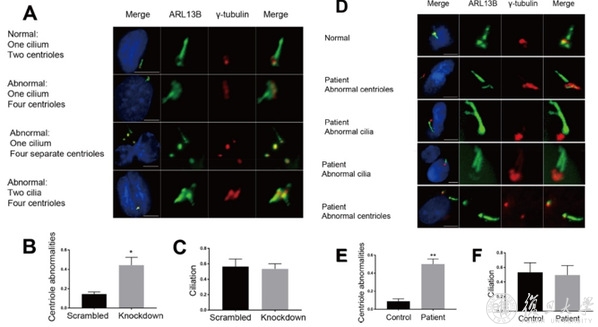
图7. 细胞实验证实,敲低ZFYVE19可以使细胞形成异常数目和形态的中心粒及纤毛
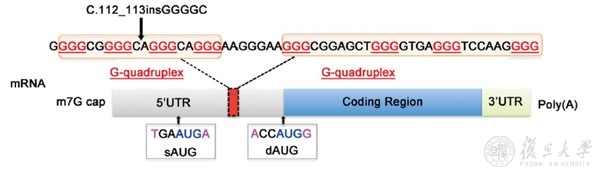
图8. ZFYVE19的mRNA结构重新分析发现以往数据库中注释蛋白翻译起始位点错误。sAUG:注释翻译起始位点。dAUG:实际翻译起始位点
栾维莎博士后、郝陈指博士和黎佳琪硕士为该论文共同第一作者,王建设教授为通讯作者。丰硕的成果来源于长期的努力,王建设教授带领的团队,从2003年开始就专注于儿童胆汁淤积症的临床和研究,且在国际上较早在病人中应用二代测序方法进行已知疾病的诊断和未知疾病的发掘,在多年临床和科研工作中积累了丰富的临床病例和研究数据资源。在未来,依托国家儿童医学中心的大平台,复旦大学附属儿科医院儿童肝病中心将继续探索,服务于更多疑难肝病患儿,为更多患儿家庭带去希望。




